[Paper Review] MolTrans: Molecular Interaction Transformer for drug–target interaction prediction
Paper Review
Huang, Kexin, et al. "MolTrans: Molecular Interaction Transformer for drug–target interaction prediction." Bioinformatics 37.6 (2021): 830-836.
1 Introduction
Challenges
Inadequate modeling of interaction mechanism
기존의 molecular representation은 전체적인 구조를 기반으로 하지만, 실제로 interaction이 일어나는 부분은 전체에서 일부임. 또 전체 구조를 기반으로한 representation은 해석하기 어려움
Restricted to limited labeled data
Drugs, proteins 각각의 수에 비해 drug-protein pair에 대한 label은 부족함
Contributions
Knowledge inspired representation and interaction modeling for more accurate and explainable prediction
- NLP에서 Ngram과 같은 방식인 Frequent Consecutive Sub-sequence (FCS) mining을 통해 sequence를 sub-structure로 나누어서 사용
Leverage massive unlabeled biomedical data
- 많은 수의 데이터를 사용함 (당연한 얘기를..)
- transformer를 이용한 augmentation (..???????)
2 Materials and methods
2.1 Problem definition
- DTI prediction
- Drug is represented by SMILES
- Target protein is represented by a sequence of amino acids
2.2 The MolTrans methods
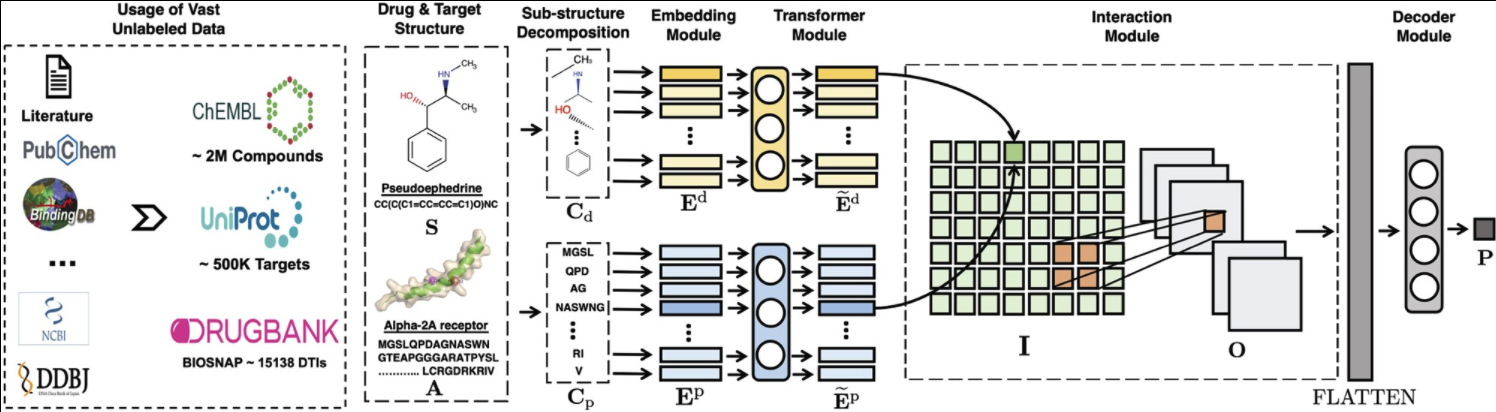
 Figure 1. MolTrans workflow
Figure 1. MolTrans workflow
2.2.1 FCS mining module

- FCS aims to generate a set of hierarchy of frequent sub-sequences for sequences.
- The significance of FCS is threefold:
- Fingerprint와 같은 encoding 방식은 intractable하지만 FCS는 discrete하고 moderate한 사이즈의 sub-structure로 explicit하게 나누기 때문에 explainable함
- Massive unlabeled data를 다루기에 적합한 방식
- Small dataset에서는 주요한 sub-structure의 frequency가 낮게 나오는 경향이 있음
- FCS can capture fundamental and meaningful biomedical semantics.
- 서로 다른 특성을 가진 데이터셋에서도 공통적으로 발견되는 sub-structure가 있었음
2.2.2 Augmented transformer embedding module
- Contextual information을 위해 transformer encoding layer를 사용
- 이 이상의 설명은 없는데.. multi-head 요소가 augmentation의 효과를 주는 것이라고 주장하는 것 같음
2.2.3 Interaction prediction module
Pairwise interaction
(수식 작성), I dimension 설명
To provide explainability, we favor dot product as the aggregation function because it generates a single scalar that explicitly measures the intensity of interaction between individual target-drug sub-structural pair. As dot product output is one-dimensional for every pair, I becomes a two-dimensional interaction map.
문제가 발생하는 지점
Neighborhood interaction
Interaction module에 CNN을 적용함
2.3 Implementation
특별한 내용이 없어서 생략
3 Result
3.1 Experimental setup
Dataset

Metrics
- ROC-AUC, PR-AUC, sensitivity (F1 score가 가장 높은 threshold 사용)
Evaluation strategies
- Training: Validation: Test = 7:1:2
- 5 x random split
3.2 Baselines
- LR, DNN, GNN-CPI(), DeepDTI(), DeepDTA(), DeepConv-DTI()
3.3 Q1: MolTrans achieves superior predictive performance

3.4 Q2: MolTrans has competitive performance in unseen drug and target setting
we randomly select 20% drug/target proteins and all DTI pairs associated with these drugs and targets as the test set.

3.5 Q3: MolTrans performs best with scarce data
We trained each method on 5%, 10%, 20% and 30% of dataset and predict on the rest of them (we use 10% of the test edges as validation set for early stopping)
3.6 Q4: MolTrans is robust in various protein families
 we test on the predictive performance on four of the largest druggable targets: enzymes, ion channels, G-protein-coupled receptors (GPCRs) and nuclear receptors. We retrieve one test set of BIOSNAP and map the target proteins to the four protein families using GtoPdb database
we test on the predictive performance on four of the largest druggable targets: enzymes, ion channels, G-protein-coupled receptors (GPCRs) and nuclear receptors. We retrieve one test set of BIOSNAP and map the target proteins to the four protein families using GtoPdb database
3.7 Q5: MolTrans allows model understanding
Interaction map에서 threshold를 통해 0/1로 나누면 1은 interaction과 관련된 sub-structure의 pair
 근데 이게 진짜 이상함. 후술하겠지만 패딩은 빼버리고 보여주는데.. 패딩을 포함하면 이상하게 나옴
근데 이게 진짜 이상함. 후술하겠지만 패딩은 빼버리고 보여주는데.. 패딩을 포함하면 이상하게 나옴
3.8 Q6: Ablation study
다소 뻔한 얘기라서 생략
4 Conclusion
MolTrans 짱 좋다고 주장
