※ 원문: HBC training 기반으로 작성하였습니다.
RNA-sequencing 데이터를 얻고 Differentially expressed genes (DEGs) analysis 을 하고자 할때, 앞서 수행해야하는 것이 정규화(normalization)이다.
관심이 없는 요소들 (Uninteresting factors) 는 최대한 배제하고, 관심 있는 생물학적 신호 (그룹 간 유의하게 차이나는 유전자 발현) 를 잡기 위함이다.
관심이 없는 요소 에는 대표적으로는 아래와 같은 요소들이 있다.
Uninteresting factors
1. Sequencing depth (시퀀싱 뎁스)
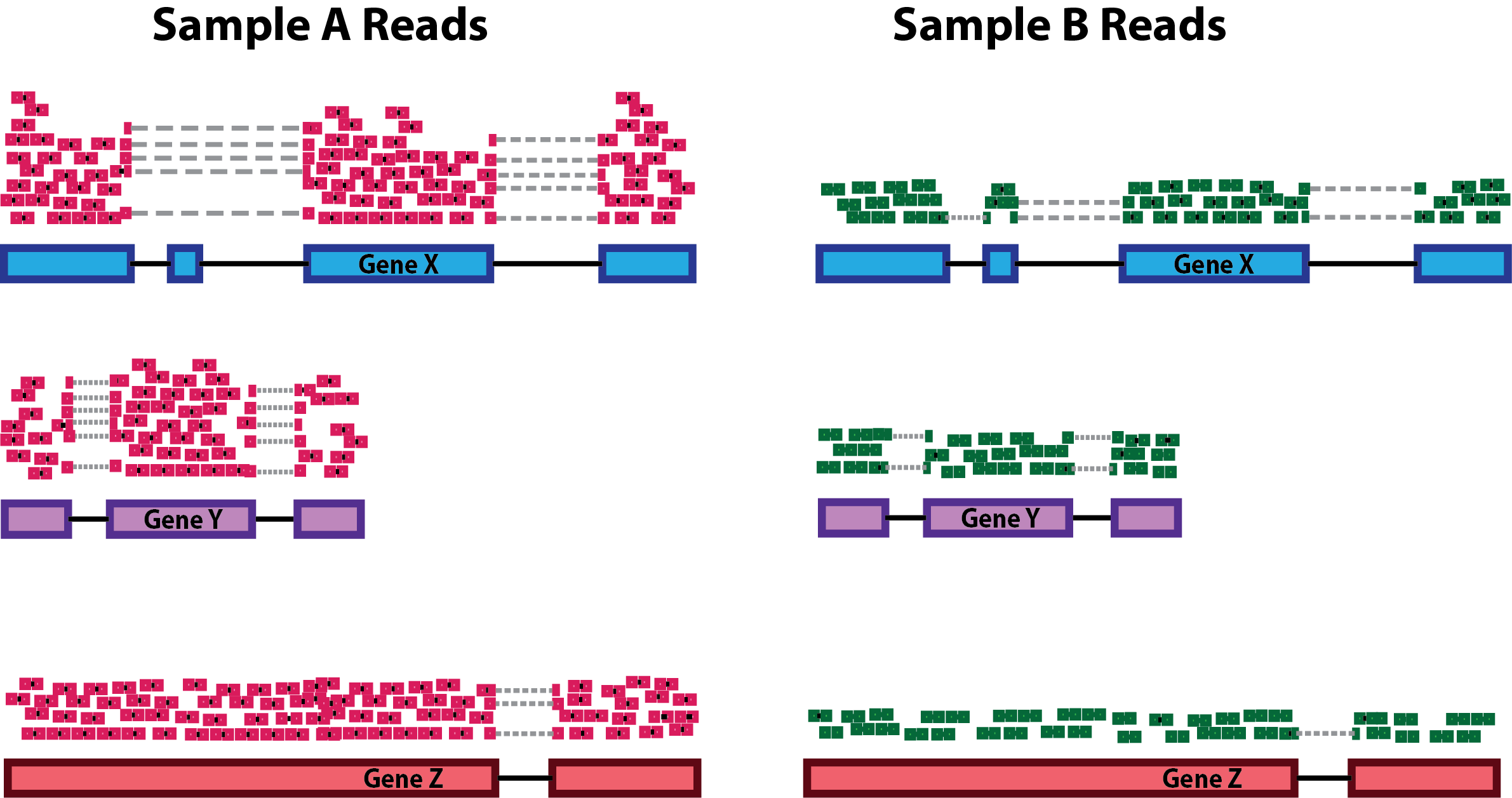 샘플간 비교 시 반드시 고려해야 한다. 예를 들어, Sample A가 Sample B보다 두 배 깊게 시퀀싱 되었다면, 같은 유전자라도 mapped reads 수가 두 배 많을 것이다.
샘플간 비교 시 반드시 고려해야 한다. 예를 들어, Sample A가 Sample B보다 두 배 깊게 시퀀싱 되었다면, 같은 유전자라도 mapped reads 수가 두 배 많을 것이다.
2. Gene length (유전자 길이)
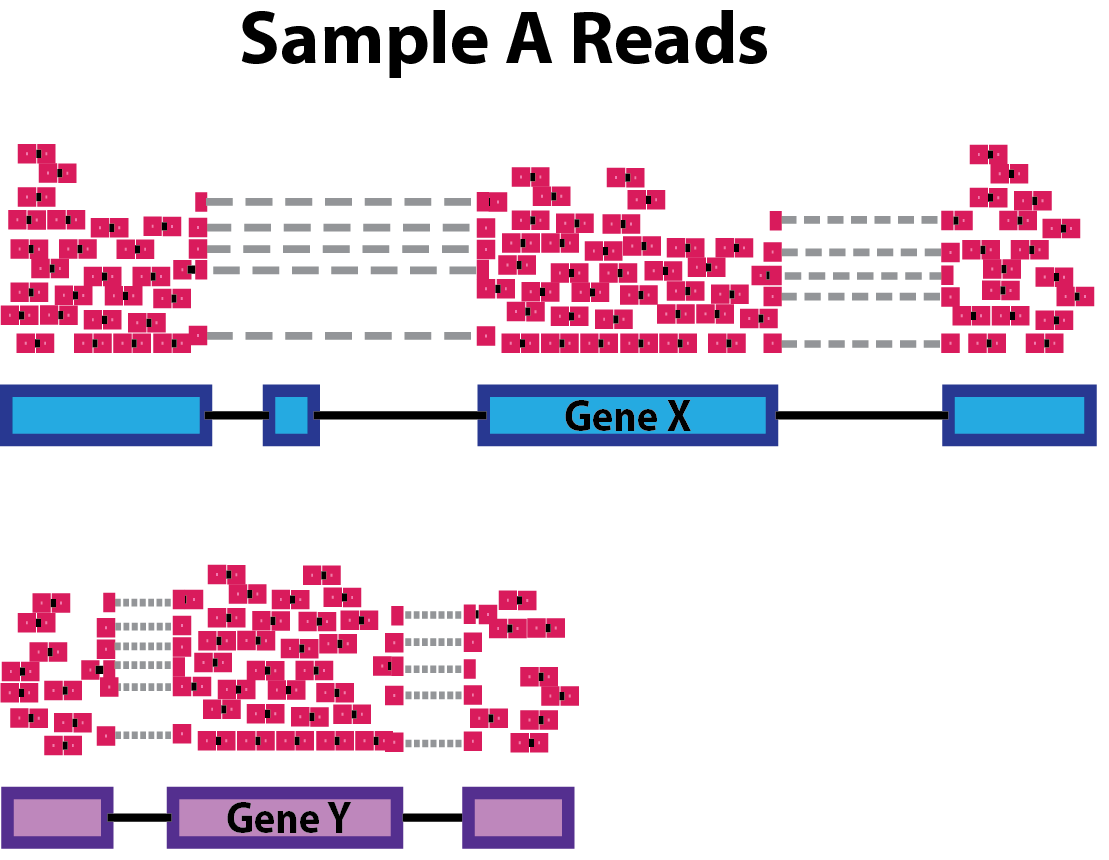
같은 샘플 내에서 유전자간 발현량을 비교할 때 고려해야 한다. 유전자 X와 Y의 발현량이 비슷하더라도, 유전자 X가 Y보다 길다면 더 많은 reads가 mapping될 것이다.
3. RNA composition
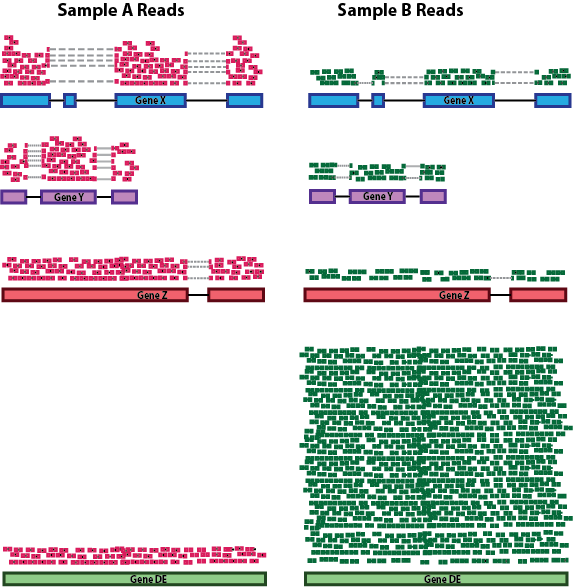
매우 높게 발현되거나 낮게 발현되는 유전자가 일부 존재하면, 정규화 방법에 따라 다른 유전자들의 발현량 계산에 영향을 줄 수 있다.
가령 어떤 암 조직 샘플에서 소수의 유전자가 정상 조직에 비해 매우 높은 발현량을 보인다고 가정해 보자. 두 조직 간 비교에서 이 유전자들이 차지하는 read count 비중이 크게 달라지게 된다.
| 정상 조직 | 암 조직 | |
|---|---|---|
| 유전자 A | 100 | 500 |
| 유전자 B | 200 | 1,000 |
| 유전자 C | 300 | 2,000 |
| 나머지 유전자들 | 99,400 | 96,500 |
| 총 read count | 100,000 | 100,000 |
위 예시처럼 암 조직에서 유전자 A, B, C의 발현량이 크게 증가하면, 상대적으로 나머지 유전자들의 read count 비중은 감소하게 된다. 이런 상황에서 단순히 각 샘플의 library size (총 read count)만을 이용해 정규화를 하면, 나머지 대다수 유전자들은 암 조직에서 과소 발현된 것 처럼 보일 수 있다.
반면 DESeq2나 EdgeR 같은 방법은 이런 bias를 줄이기 위해, 어떤 그룹의 샘플이든 대부분의 유전자 발현량이 일정하다는 가정 을 바탕으로 정규화 과정을 거친다. 각 유전자의 발현량 중앙값 (DESeq2) 혹은 trimmed mean (EdgeR)을 이용해 normalization factor를 계산함으로써, 소수 유전자의 극단적인 발현량 변화에 의한 영향을 최소화하는 것이다.
Normalization methods
이러한 "관심이 없는" 요소를 제거 또는 보정해주는 정규화 방법에는 여러가지가 있다.
CPM (counts per million)
각 샘플의 유전자 read count를 샘플의 총 read 수로 나눈 뒤 1,000,000을 곱해준다. 이를 통해 sequencing depth를 보정한다.
(: 유전자 의 read count, : 해당 샘플의 총 유전자 수)
CPM은 library size (=sequencing depth)만 고려할 뿐 유전자 길이는 고려하지 않기 때문에, 서로 다른 유전자 간 발현량 비교에는 적합하지 않다. 또한 technical bias나 RNA composition의 영향을 많이 받을 수 있어 정확한 DEG 분석이 어려울 수 있다.
원 링크에서의 표현은 아래와 같다.
gene count comparisons between replicates of the same sample group. NOT for within sample comparisons or DE analysis
TPM (transcripts per kilobase million)
CPM과 유사하지만, 유전자 길이까지 고려한다. 우선 각 유전자 read count를 유전자 길이(kb)로 나눠준다. 그 후 이 값을 샘플의 총 값으로 나눈 뒤 1,000,000을 곱해 sequencing depth와 gene length 모두 보정한다.
(: 유전자 의 read count, : 유전자 의 길이(kb), : 해당 샘플의 총 유전자 수)
TPM은 유전자 길이를 반영해 같은 샘플 내 서로 다른 유전자 간 비교는 가능하다. 하지만 여전히 RNA composition bias가 해결되지 않아 샘플 간 비교나 DEG 분석에는 한계가 있다.
원 링크에서의 표현은 아래와 같다.
gene count comparisons within a sample or between samples of the same sample group; NOT for DE analysis
RPKM/FPKM (reads/fragments per kilobase of exon per million reads/fragments mapped)
TPM과 유사하게 sequencing depth와 gene length를 보정한다. TPM과 거의 같은 방식이지만, scaling factor가 달라 정규화 후에도 샘플간 total read count가 다르다. 따라서 샘플 간 비교에는 부적합하다. (: 유전자 의 read count, : 유전자 의 길이(kb), : 해당 샘플의 총 유전자 수)
또한 RPKM/FPKM 역시 RNA composition bias 문제가 남아있어, 정확한 DEG 검출에는 적합하지 않다.
원 링크에서의 표현은 아래와 같다.
gene count comparisons between genes within a sample; NOT for between sample comparisons or DE analysis
따라서 CPM, TPM, RPKM/FPKM 모두 기본적인 sequencing depth나 gene length 보정은 해주지만, DEG 분석에 필수적인 RNA composition bias는 해결하지 못한다. 때문에 DEG 검출을 위해서는 DESeq2나 EdgeR 같은 방법을 사용하는 것이 바람직하다. 이들은 normalization factor 계산시 대다수 유전자의 발현량이 안정적이라는 가정 을 바탕으로, 소수 유전자의 편향된 발현량 변화의 영향을 최소화한다. 따라서 실제 생물학적 변이에 의한 DEG를 보다 정확히 검출해낼 수 있게 해준다.
DESeq2's median of ratios
sequencing depth와 RNA composition을 보정하기 위해 geometric mean 대비 각 유전자 count의 ratio 중앙값을 사용한다. 이는 그룹 (조건)이 달라도 대부분 유전자의 발현량은 변하지 않는다는 가정하에 편향된 값을 보정 하는 방식으로, DEG 분석에 적합하다.
- 각 유전자의 geometric mean (기하 평균) 을 샘플들에서 계산하여 pseudo-reference를 만든다.
- 각 샘플의 유전자 read count를 pseudo-reference로 나눠 ratio를 구한다.
- 한 샘플내 모든 유전자의 ratio 중앙값을 size factor로 정한다.
- Raw read count를 size factor로 나누어 정규화한다.
원문 링크엔 토이 데이터를 이용해 시연하는 과정까지 있으므로 한번 직접 따라가보는 걸 추천한다.
마치며
이처럼 각 정규화 방법은 장단점이 있기에 실험 디자인과 목적에 맞게 사용되어야 한다. 일반적인 RNA-seq의 DEG analysis 에서 가장 많이 쓰이는 건 DESeq2나 EdgeR의 방법이다. 각 실험 조건에 맞는 정규화를 통해 관심있는 생물학적 변이를 제대로 관찰할 수 있도록 하는 것이 중요하다. 물론, RNA composition bias를 무시하고, TPM, FPKM, CPM (혹은 심지어 raw count)로 DEG 결과를 제시하는 논문도 있다.
샘플의 수가 많다면 technical bias를 어느 정도 상쇄할 수 있어 재현/검증 가능한 결과를 얻을 수 있다. 하지만 가능하다면 최대한 이런 bias는 사전에 제거하는 것이 false discovery 를 예방하는 데 도움이 될 것이다.
사족(1): DEG 분석에서의 가정, 귀무가설과의 연관성
지난 DEG 포스팅 에서 언급한 바와 같이, DEG 분석에서 제일 중요한 가정은 "대부분의 유전자는 두 그룹 간 발현량 차이가 없다" 는 귀무가설이다.
이 가정은 DESeq2와 EdgeR의 정규화 방법에도 반영되어 있다.
- DESeq2의 median of ratios 방법은 모든 샘플들의 각 유전자 발현량 기하평균(geometric mean)으로 pseudo-reference sample을 만들고, 각 샘플의 유전자 발현량을 이 reference로 나눈 비율의 중앙값을 size factor로 사용함.
- EdgeR의 TMM (Trimmed Mean of M-values) 방법은 한 샘플을 reference로 정하고, 다른 샘플들과의 M(log ratio)값 분포에서 극단적인 값들을 제외한 후 평균을 normalization factor로 사용함.
두 방법 모두 대다수 유전자의 발현량은 샘플 간 차이가 크지 않을 것이라는 가정하에, 소수 유전자의 극단적인 발현량 변화가 미치는 영향을 최소화하고자 한다.
이는 곧 대부분의 유전자는 그룹 간 발현량 차이가 없다는 DEG 분석의 귀무가설과 일맥상통한다.
따라서 DESeq2와 EdgeR은 이러한 귀무가설을 전제로, 실제 biological variation에 의한 DEG를 보다 민감하게 검출할 수 있는 것이다. 반면 CPM, TPM, RPKM 등의 방법은 이런 가정을 반영하지 않아, DEG 검출력이 상대적으로 떨어질 수 있다.
사족(2): scRNA-seq
한편, single cell RNA-seq 중 제일 많이 쓰이는 10X Genomics 방식은 mRNA poly-A tail 부분에 primer를 붙여 3' 쪽 서열만을 읽어내는 방식이다. 이는 UMI(Unique Molecular Identifier)라는 짧은 핵산 서열을 함께 부착함으로써, PCR duplicate를 제거하고 절대적인 mRNA 분자 수를 계량화 (Qunantification) 할 수 있게 해준다.
이러한 3' 쪽 서열만 읽는 것은 transcript의 길이에 따른 bias를 최소화한다는 장점이 있다. 때문에, 10X scRNA-seq 데이터 분석 시에는 RPKM/FPKM/TPM 대신 gene length 보정이 필요 없는 CPM (counts per million)이 자주 사용된다. 각 cell의 UMI count를 cell 내 총 UMI count로 나눈 후 1,000,000을 곱해주는 방식이다. 이는 각 cell의 library size를 보정함과 동시에 counts를 백만 분율로 표준화한다.
하지만 CPM은 여전히 각 cell의 RNA 구성비 차이에 의한 bias 가능성을 내포하고 있다. 앞서 언급한 예시와 같이, 특정 유전자가 과도하게 높은 발현량을 보이는 cell이 존재한다면, 그 cell 내 다른 유전자들의 CPM 값은 과소 추정될 수 있다. 이를 해결하기 위해, scRNA-seq에서도 bulk RNA-seq과 마찬가지로 DESeq2, EdgeR 등의 방법을 적용해볼 수 있겠다.
scRNA-seq 데이터의 정규화에 대한 golden standard는 없고, 여전히 도전적인 과제로 남아있다. CPM, DESeq2, EdgeR 등 기존의 방법을 우선 시도해보되, 데이터의 질과 분석 목적에 따라 다양한 접근을 시도해볼 필요가 있다. 사용한 방법에 대한 명확한 근거를 제시하고, 가능하다면 여러 방법을 비교 검증하는 것도 중요하다.

매 번 이해가 잘 되게 잘 설명해주시는 것 같아요. 감사합니다.
이번에도 한 가지 질문 드리고자 하는 건 조금 다른 내용일 수 있지만, bulk에서 single cell 단위로 넘어가면서 단순히 bulk 단위로 봤을 때의 limitation을 극복할 수 있게 된 것 같이 spatial로 보는 것은 연구적인 측면에서 어떠한 이점이 있는 건가요?